"The sustained dependability of a tireless heart relies…on the performance of the trillions of chemical reactions occurring in its aggregate of cells every instant of its function."
Sherwin Nuland, MD, Author of The Wisdom of the Body, 1997
Under a microscope, the true grandeur of the heart reveals itself.Magnified, a rod-shaped heart muscle cell taps out a constant beat. A closer look within the cell reveals a series of thin contractile fibers called myofilaments that are the machinery driving these contractions. In the left ventricle alone, there are nearly 5 billion of these cells beating rhythmically, as if they are all listening to the same snappy tune.
The chemical chain of events that underlies the beating of these cells and of the heart as a whole is truly remarkable. First, there’s the electrical impulse along the cell’s membrane; then channels open in the cell membrane allowing sodium to flow into the cell. After that, more channels open and calcium enters and binds to a tiny structure near the membrane; then, much more calcium explodes out of that structure into the cell’s inner fluid and combines with a myofilament protein called troponin. Troponin then changes shape to allow two other proteins, actin and myosin, to come together. The joined proteins slide past each other in such a way as to shorten the cell, pulling the ends of the cell inward—this is the actual contraction—and then the whole process reverses itself as the heart relaxes in preparation for the next beat.
 |
|
Magnified, a rod-shaped heart muscle cell (myocyte) contracts its myofilaments. Billions of these cells contracting in synchronization produce a heart beat. |
During the past 30 years, scientists have made some intriguing discoveries about the process that changes an electrical impulse into a muscle contraction. These discoveries have led to novel hypotheses about aging and disease. Scientists have found that age-related changes in heart muscle cells (myocytes) help explain alterations in the heart as a whole. For instance, they’ve learned there are fewer myocytes to do the work as we age and those that remain enlarge, compromising their ability to pump blood efficiently. They’ve also discovered much about how these changes could interact with disease processes and found clues to how exercise affects the biochemistry of cells. Scientists have begun to question some of the long-held theories about the nature of the aging heart, including whether some myocytes can replicate and what role aging may have in this process. And they’ve learned a great deal more about the critical role calcium plays in the drama of the aging heart.
The Marvelous Calcium Pump
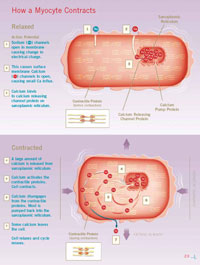
Scientists have long known that calcium—the mineral that helps keep your teeth and bones strong—also has an important job within your heart. Calcium entering the myocyte’s inner fluid or cytosol binds with other contractile proteins to bring about contraction. Calcium leaving the cytosol allows the cell to relax. It’s this constantly changing ebb and flow of calcium in and out of the cytosol of heart muscle cells that is the essence of the heart beat. (See How a Myocyte Contracts.)
At the beginning of the calcium cycle—which coincides with the heart filling with blood—calcium in the cytosol and surrounding the contractile filaments of each myocyte is at least 10,000 times lower than calcium levels in your blood and in other fluids between your cells, called the intercellular spaces. As the cycle progresses, pores (or channels) in cell membranes open and close, allowing various salts to flow in and out of the cell. This activity triggers momentary fluctuations in the positive and negative electrical charges across the cell’s membrane. When these fluctuations reach a particular threshold, an electrical discharge occurs. This discharge, called the action potential, essentially flips a switch on the myocyte’s membrane to open pores that allow a small amount of calcium to enter the cell.
 |
|
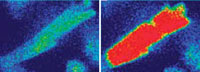
Between heart beats,calcium levels (red) diminish in a myocyte’s inner fluid or cytosol,allowing it to relax (left).During con-traction,a large amount of calcium surges out of a myocyte’s sarcoplasmic reticulum,right,triggering tightening or shortening of the cell’s muscle fibers (right). This process occurs almost simultaneously in every cell in the heart wall,causing them to contract and pump blood out of the heart. |
This tiny bit of calcium binds to openings called calcium release channels on the sarcoplasmic reticulum, an organelle (a small cellular “organ”) that serves as a storage bin for calcium. In response, the sarcoplasmic reticulum releases a large amount of its stored calcium into the cell. The calcium released from this storage compartment binds to the cell’s myofilaments, causing them to tighten or shorten. As the myofilaments tighten, the myocyte compresses (shrinking in length and fattening in width). This process occurs almost simultaneously in every cell in the heart wall, causing them to contract and pump blood out of the heart.
In order for the heart to relax, the cycle winds down and calcium detaches from the myofilaments. A cellular mechanism kicks in and pumps most of the calcium back into the storage bins located in the sarcoplasmic reticulum. Any residual calcium is driven out of the cell through specialized exit calcium carriers located on the cell’s membrane. These carriers are proteins that swap calcium inside the cell for sodium outside of it. Then the cycle restarts in preparation for the next heart beat.
If this system fails, and calcium cycling gets out of whack, chaos can ensue. The heart, for instance, can’t relax and fill with blood properly and diastolic pressure in the heart increases. In addition, individual cells may fire off rapidly and independently, resulting in arrhythmias—variations from the normal heart beat rhythm—and fibrillation, which is a very rapid twitching of individual muscle fibers. In particular, older hearts are more susceptible to spontaneous calcium oscillations than younger hearts, and it takes fewer oscillations to bring about fibrillation. Fibrillation in the left ventricle leads quickly to acute heart failure and to death if not treated.
Heart failure occurs when the heart loses its ability to pump enough blood to meet the body’s requirements. In particular, heart failure causes the heart to gradually lose its reserve pumping capacity and work less efficiently. Blood pressure and flow to body organs drops. The kidneys sense this and send out signals prompting retention of body fluid, which contributes to swelling. This can cause a backup of fluid into the lungs and body tissues triggering shortness of breath, swelling of the legs and feet, and other symptoms. As heart failure progresses the effects can become quite severe, and patients often lose the ability to perform even modest physical activity. Eventually, the heart's reduced pumping capacity may interfere with routine tasks, and individuals may become unable to care for themselves. Heart failure rises exponentially with advancing age, and studies of the calcium cycle in heart cells suggest a number of possible reasons.
|
How a Myocite Works |
 |
|
|
When a Good Pump Goes Bad
Imagine sitting calmly in a living room chair when the smoke detector goes off. As you scramble to quickly get out of the house, your heart starts beating faster. A few moments later, after you discover it was a false alarm, you return to your comfortable chair, and your heart rate slows again. As this scenario suggests, your heart beat can vary from moment to moment. And your heart’s ability to respond to these changes depends a lot on calcium. The more calcium your heart cells release from their intracellular storage bins, the greater the force of the heart’s contractions. But how well these mechanisms work depends on how much calcium can be pumped from these storage bins between heart beats. In young hearts, these calcium pumps work quite well, but in older hearts these pumps are much less efficient.
After the heart beat, if you recall, most of the calcium returns to the storage bins in the sarcoplasmic reticulum and then awaits the next signal to do its job again. Scientists began taking a closer look at this mechanism when they learned that muscle from older hearts takes longer to relax than muscle from younger hearts. One of the prime suspects for this phenomenon was calcium. To test this idea, Dr. Lakatta and his colleagues at the NIA used a protein that binds to calcium and gives off a blue light to detect how much calcium is in a cell at any one time. When they injected this calcium-sensing protein into myocytes within heart muscle in laboratory dishes, the blue light showed that calcium levels, after a contraction, fell more slowly in older myocytes. Or, putting it in biologists’ terms, the cytosolic calcium transit was longer. But why? Could the calcium be spending longer in the inner fluid because the sarcoplasmic reticulum wasn’t removing it as quickly in older cells?
The answer was yes. In experiments, NIA scientists isolated the sarcoplasmic reticulum from the rest of the heart cell, placed it in a test tube, and then added calcium. The sarcoplasmic reticulum took up the calcium more slowly in samples from older animals than those from younger ones.
Subsequent studies confirmed that the sarcoplasmic reticulum—or more precisely, a protein on this organelle—removes calcium more slowly in older hearts. Researchers have found that older cells have lower amounts of this particular protein, often called the calcium pump protein because it removes the calcium in a series of repeated movements. In essence, the sarcoplasmic reticulum removes calcium from the inner fluid more slowly in older hearts because there are fewer pumps, and those that remain don’t work as well because of communication breakdowns between the brain and the heart.
If these pumps aren’t working properly or have shut down, the sarcoplasmic reticulum won’t fill as well as it should with calcium, and there won’t be enough calcium to fulfill the heart cells’ needs, particularly during exercise or stress.
Once scientists learned about the pump protein, the next question was about that protein’s gene. Proteins make up a huge category that includes enzymes, growth factors, hormones—almost all the substances that are responsible for the day-today functioning of living organisms. Proteins are produced by genes in the nucleus of every cell. Each protein has its own gene. Cells translate gene codes into proteins through a complex, multistep process called gene expression. Any alteration in this process can lead to changes in the end product, the protein.
In the case of the pump protein, the gene that produces it is only about half as active in older hearts as in younger hearts. The end result of all of these changes is a decline with age in the maximum strength of the heart beat during strenuous activity. Reduced calcium pumping also prolongs the time it takes for heart cells—and in turn, the heart as a whole—to return to a relaxed state. As a consequence, the heart can’t fill with blood as readily as it once did and prepare for the next heart beat.
Like other changes, the longer calcium transient appears to be one way that the heart adjusts to age, or more specifically to the stiffer arteries that accompany aging. Unfortunately, like those other changes, this adjustment also has a cost.
“It makes sense from an engineering standpoint to have a longer contraction if you’re pumping blood into stiffer vessels,” Dr. Lakatta says. “The downside is that when you alter the dynamics of calcium, various stresses can more easily throw the calcium out of balance. One consequence of this is that an older person is more apt to feel short of breath during vigorous exercise.”
Age Lengthens Action Potential
In addition to calcium transit, two other clusters of events in myocytes seem to be affected by age. One is the action potential. This is a transient alteration in the amounts of positive and negative charges on either side of the myocyte membrane. As mentioned earlier, the action potential triggers the opening of sodium and then calcium channels in the membrane.
The action potential is prolonged in older hearts and may contribute to the longer calcium transient. This occurs because as the heart ages, there are coordinated declines in both the activity and number of proteins involved in the action potential as well as the proteins that respond to its signals. A longer action potential generates a longer calcium transit, which in turn, produces a longer contraction. Each of these processes is controlled by specific proteins.
The prolonged action potential helps older hearts work well in most situations. It does this in two ways. First, pores on the myocyte’s membrane stay open longer to allow more calcium to enter the cell between beats. Second, the proteins that carry calcium out of the cell and sodium back in work more slowly. The net result is that more calcium is available within the cell. These effects allow the weaker sarcoplasmic reticulum—which has fewer pumps—to load up on calcium in preparation for the next beat. But these adjustments, like so many other cardiovascular adaptations, may have a downside. For instance, in an aging heart the long action potential adaptation works well at slower heart rates. But during a rapid heart rate, the longer action potential contributes to calcium dysregulation of myocytes. As a result, the older heart doesn’t respond as dynamically to the needs of the body as a young heart. So, a prolonged action potential is yet another possible reason that an older person usually can’t do as much exercise as someone younger.
Contractile Proteins
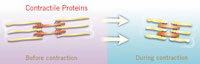
The other mechanisms that change with age involve contractile proteins—actin, myosin, troponin, and others—that interact to shorten, or contract, the myocyte. These contractile proteins pass through a series of steps, triggered by calcium, which bring actin and myosin together into crossbridges. The crossbridges use energy released during the transaction to shorten the cell. With age, one part of the crossbridge alters—the part called the myosin heavy chain.
The myosin heavy chain can be produced in two slightly different forms, one dubbed alpha, the other beta. In experimental animals, the alpha myosin heavy chain decreases with age, while the beta increases. The same seems to be true in the human atrium. When the proportion of alpha myosin heavy chain is reduced in isolated cells, the contraction speed is slower.
Changes in the myosin heavy chain have been traced back to the genes involved—alpha is expressed less with age, beta more. The expression of these genes is regulated by proteins called transcription factors that start or regulate the first steps of cellular reproduction. One of the transcription factors for the myosin gene is the same as that for the sarcoplasmic reticulum pump, suggesting that there is a common aging link between the two cellular mechanisms. Studies in rodents suggest the activity of these factors declines with age. Because of these changes the expression of genes in the aging heart tends to go back to patterns of gene expression seen in the fetus.
These age-related changes in myosin and other contractile proteins, in conjunction with alterations in calcium transit and action potential, actually help the older heart work more efficiently. That’s because slower and longer contractions don’t use as much energy. Prolonged contractions also allow the older heart to eject blood into the arteries later in the heartbeat. This adaptation is good because it improves the blood flow through an older person’s stiffer arteries.
Free Radical Damage
Myocytes produce free radicals, unstable oxygen molecules that can disrupt a cell’s inner workings. As the heart ages, these free radicals can greatly alter how well the cellular calcium pumps on the sarcoplasmic reticulum work.
In myocytes, most free radicals are produced in tiny cellular organelles called mitochondria and by an enzyme in cell membranes called NADPH oxidase. Mitochondria convert oxygen and food into an energy-releasing molecule that powers most cellular processes. But during this process they also produce potentially harmful byproducts such as oxygen free radicals. A free radical can be produced by almost any molecule when it loses an electron from one or more of its atoms. In heart muscle cells, they are commonly created when mitochondria combine oxygen with hydrogen to form water. Free radicals can cause extensive damage to proteins,membranes, and DNA. As we age, mitochondria become less efficient, progressively generating less energy releasing molecules and more free radicals.
In the aging heart, free radicals damage proteins, membranes, and calcium pumps on the sarcoplasmic reticulum that myocytes need to produce contractions. As a result of this cellular damage, myocytes can’t process calcium as well. As calcium builds up in the cell, it can begin to contract erratically, causing an arrhythmia. If this arrhythmia spreads to other cells, it can eventually disrupt beating throughout the heart and lead to serious complications.
|
Contractile Proteins |
 |
|
|
Nitric Oxide
Calcium isn’t the only factor that helps the heart respond to the body’s increased need for blood and oxygen during sustained exertion or times of stress. Nitric oxide, a potent chemical messenger that helps regulate blood flow in the arteries, also signals the heart to pump harder at critical times.
As we just learned, heart cells can increase their contraction force by releasing more calcium from the sacroplasmic reticulum. Once released from this cellular storage bin, calcium binds to troponin and other contractile proteins, which trigger the heart beat. The more calcium that binds to these contractile proteins, the greater the force of the contraction. But there’s at least one other pathway, the Frank-Starling mechanism, that can increase the force of these contractions. This mechanism kicks in when increased blood flow into the heart stretches myocytes, signaling them to contract harder and produce more force. Normally, these two pathways work independently to tap the heart’s reserve capacity. But if cell stretch is sustained for prolonged periods, the amount of calcium released into heart cells during contractions gradually increases. This suggests that some coordinating mechanism is operating to ensure that, even under duress, the heart continues to pump enough blood to the body. But what this link might be mystified scientists for many years.
Steven Sollott, MD and his colleagues at the NIA theorized that sustained cell stretch prompts an enzyme, nitric oxide synthase, to produce nitric oxide. Nitric oxide, in turn, binds to the calcium release channels in the sarcoplasmic reticulum and promotes the release of calcium into the cell.
To test this theory, the investigators conducted a series of experiments with adult rats and mice—mammals with cardiovascular systems similar to humans. Some rodents were genetically altered so they could not produce the enzyme that makes nitric oxide. Others were given drugs that blocked the production of the chemical messenger. In both cases, sustained cell stretch no longer triggered increased calcium release in heart cells. Previous studies have shown that nitric oxide levels may be diminished in heart failure and other cardiovascular diseases.
“Knowing that this nitric oxide mechanism exists and how it functions in the normal heart may help us understand what happens to it with age and disease,” Dr. Sollott says. “This discovery offers scientists an opportunity to consider whether therapies that sustain or enhance the functioning of this mechanism might help aging hearts stay healthy and continue working properly.”
New ideas are also emerging about two other phenomena that have puzzled gerontologists and cardiovascular scientists for decades. As heart cells get older, there are fewer of them to do the necessary work, and those that remain get bigger.
Bigger Heart Cells...
To efficiently pump blood, the stiffness of the heart changes as it beats. When it is filling, the heart needs to be as relaxed as possible to allow blood to freely flow into it. When it contracts, it stiffens so that the pressure it exerts is greater than that found in the arteries. When this happens, blood is forced into the arteries with a minimal amount of effort. If there is a load mismatch—meaning the force differential between the heart and arteries isn’t very good—the heart can’t empty as well as it once did. As a result, the heart has to work harder to get blood into the arteries. If this occurs on a regular basis, some heart cells die, others enlarge, and the heart walls thicken.
|
Opposing Pressures |
 |
|
|
This problem gradually increases as we get older. In some cases, the cells that remain enlarge up to 40 percent. The enlargement of these remaining myocytes seems to be the principal mechanism for the thickening of the heart walls—the hypertrophy—that occurs with normal aging.
Much evidence suggests that myocyte enlargement and the consequent thickening of the heart walls are ways that the heart adjusts to increased loads, especially from the growing stiffness of the arteries. Extra loads also may develop as the result of disease.
One reason cardiovascular researchers are so intrigued by myocyte enlargement is because of its possible links to disease. While enlargement seems to occur in response to aging and stiffening of the arteries, it is exaggerated by disease, such as coronary artery disease and high blood pressure. (However, enlargement occurs with high blood pressure at any age).
While myocyte enlargement seems to be one way that the heart adapts to increased loads, there is also evidence that at the oldest ages, it no longer adapts as much. Older animals, for instance, have less enlargement in response to heart overloads than younger animals. This failure or slowing of the adaptive response may explain why 80-year-olds are much more likely to experience heart failure following a heart attack than 60-year-olds.
These findings are yet another clue suggesting that specific age-associated changes in healthy hearts and blood vessels compromise their ability to respond to everyday stress and strain. In turn, these changes gradually lower the heart’s resistance to certain cardiovascular conditions including left ventricular hypertrophy, atrial fibrillation, and congestive heart failure.
...But Fewer of Them
As we age, even the healthiest hearts lose cells. In a robust 70-year-old man without heart disease or high blood pressure, these age-related losses are estimated to account for up to a 30 percent reduction in the total number of myocytes in the heart. Although it’s unclear whether this loss of heart cells is good or bad for the body as a whole, evidence suggests that loss of a significant number of heart cells may contribute to the decline in cardiovascular health in older people.
Cardiovascular scientists are exploring why some myocytes die while others continue to thrive. Injury, due to a lack of oxygen or ischemia, seems to be one of the prime killers. But studies suggest that programmed cell death—apoptosis—could be a significant factor as well.
Apoptosis is a process in which a cell orders itself to stop functioning, shrink, and ultimately dissolve. It has been observed in other cells in the body, where it may be a mechanism for adjusting to development or removing unwanted or potentially dangerous cells, such as cancer cells, from the body. At least one study suggests that apoptosis in the heart becomes more common with age. And other research has found that excessive apoptosis may contribute to decline in the aging cardiovascular system.
 |
|
Christiaan Leewenburgh, PhD |
A number of molecular processes, such as increased free radical production, can activate a cell’s apoptotic or self-destruct mechanism. In rodents, for instance, NIA grantee Christiaan Leewenburgh, PhD, of the University of Florida and his colleagues found that cytochrome c, a mitochrondial protein, becomes a signal for cell death if it “leaks” from the mitochondria. The hearts of older rats released greater amounts of that cell-death signal than did the hearts of younger rats. This difference may be partly responsible for the increase in heart cell death.
Cardiovascular scientists studying apoptosis are particularly interested in the role of a process called cardiac stretch. Myocytes are connected, so when one dies—for whatever reason—others must stretch to maintain the connections. As myocytes are stretched, they release chemical substances called growth factors, such as norepinephrine and angiotensin. These growth factors may not only help explain why these cells enlarge, but also why some of them die. In laboratory dishes, for instance, the same growth factors that regulate heart cell growth also trigger apoptosis in some myocytes. However, these and other growth factors may have an equally important role in a process that was once thought to be impossible: the replication and regeneration of heart cells.
The Untapped Promise of the Aging Heart
For decades it was believed that the heart had a set number of myocytes, and once one of these cells died, they couldn’t be replaced. Because these cells were thought to be unable to divide, according to this view, their numbers progressively decreased with age and ultimately impaired heart function.
While this view was widely accepted, it was never proven. And now, compelling but controversial evidence raises new questions about its validity.
 |
|
Piero Anversa, MD |
One of the first challenges to this dogma came in 2001 when scientists from New York Medical College in Valhalla, New York found large scale replication of heart muscle cells in two regions of the heart, and identified several other key indicators of cell regeneration. These scientists, led by NIA-grantee Piero Anversa, MD studied myocytes from the hearts of 13 patients, 4 to 12 days after their heart attacks, and from the hearts of 10 patients who did not have cardiovascular disease. Samples were obtained from the border zone near the site of the heart attack and from a more distant site from the damaged tissue.
By viewing these areas of the heart with a high resolution confocal microscope, the investigators were able to measure the expression of a protein found in the nucleus of dividing heart muscle cells. This protein is expressed during all phases of a cell’s life cycle and is a strong indicator of ongoing cell division.
The scientists also obtained images of cell division and found other evidence of myocyte replication, including the formation of the mitotic spindle, and contractile ring, critical structural indicators of cell division. In comparison with normal hearts, the number of myocytes multiplying in diseased hearts was 70 times higher in the border zone and 24 times higher in the remote tissue.
But where were these new hearts cells coming from? Subsequent studies—in both animals and humans—suggested that, although most adult heart cells probably aren’t able to replicate, a small core of adult stem cells might exist in the heart or in the bone marrow that are capable of replenishing and replacing damaged or dying myocytes. However, other scientists have had difficulty duplicating these results. So for now, the notion that adult stem cells can regenerate heart muscle remains a tantalizing, but unproven, possibility.
Some cardiologists theorize that adult stem cells in young hearts might be able to produce enough new myocytes to replace those that are naturally dying. In effect, these stem cells might help keep the total number of myocytes stable in the young heart. But mounting evidence suggests that even if these stem cells exist, their numbers decline with age. As a result, some scientists suspect there are fewer new heart cells to replace older ones which are dying in greater numbers due to age, injury, and other problems. So the deterioration of the older heart could be related, in part, to the inability of aging cardiac stem cells to replace dead and dying myocytes with new ones.
Gerontologists exploring this exciting new area of research have many questions, but few answers at this point. For instance, does the number of cardiac stem cells really decline with age? What role do growth factors play in regulating the activity of these cells? How are the changes in cardiac stem cells linked to other age-related alterations in the heart and arteries? Why are scientists having so much difficulty replicating the earlier findings? Are the myocytes in the heart of an 80-year-old basically the same ones present in his or her heart at birth or have the cells been gradually replaced over the years like the skin? And perhaps most importantly, if these stem cells exist, can anything be done to stimulate them to produce new myocytes that will counteract the effects of age in the older heart?
The key to answering this final question lies in learning more about stem cells and how they work. Investigators already know that stem cells have important characteristics that distinguish them from other types of cells. Unlike most cells in the body, such as skin or brain cells, which are dedicated to performing a specific function, stem cells are not specialists. But under certain physiologic or experimental conditions, they can be induced to become cells with special functions such as the beating cells of the heart muscle or the insulinproducing cells of the pancreas. Another unique characteristic of stem cells is their ability to renew themselves for long periods through cell division.
Scientists primarily work with two kinds of stem cells from animals and humans: embryonic stem cells and adult stem cells.Most of the basic science research discoveries on embryonic and adult stem cells come from research involving animals, particularly mice. Embryonic stem cells are derived from embryos. Specifically, embryonic stem cells are derived from embryos that develop from eggs that have been fertilized in vitro.
 |
|
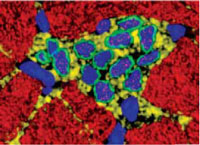
In this microscopic image,adult myocytes (red) surround a cluster ofstem cells (blue) growing to maturity in the heart. Many questions remain about these cells and their potential role in the aging heart. |
Adult stem cells typically generate the cell types of the tissue in which they reside. A blood-forming adult stem cell in the bone marrow, for example, normally produces many types of blood cells such as red blood cells, white blood cells, and platelets. Until recently, it had been thought that a bloodforming cell in the bone marrow—which is called a hematopoietic stem cell—could not give rise to the cells of a very different tissue, such as myocytes in the heart. However, a number of experiments over the last several years have raised the possibility that stem cells from one tissue may be able to make cell types of a completely different tissue, a phenomenon known as plasticity.
Because of this flexibility, stem cells hold enormous potential for cell replacement or tissue repair in heart disease and many other age-associated disorders. Gerontologists are seeking to find out if these cells will yield any practical interventions that might promote healthy aging.
“We’ve made substantial progress, but there is a lot more to be learned,” according to Dr. Lakatta. “Finding ways to activate these cells and get them to where they are needed in the heart and ensuring that they develop into heart cells are significant challenges.”
While daunting, these and other challenges are motivating gerontologists to investigate many new interventions that in the future could help keep aging hearts and arteries healthy.
As we age, for instance, pressure increases in the arteries, and this can affect the structure and function of the left ventricle. In fact, a growing number of scientists suspect that age-related changes in the blood vessels may actually instigate many of the transformations that occur in the older heart.
<< Back | Next >>

