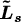 in
in
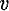 s = 1 States
s = 1 States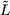
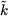 L⟩ transform according to the
analogous equations (eq. 33) and
(eq. 48), it is possible to use standard
vector coupling techniques [22] to obtain linear
combinations of products of the form |k′Jm⟩
| ′L⟩ which correspond to wave
functions characterized by the quantum numbers
|J m R kR⟩. The two new quantum
numbers R and kR, which replace k and
L, are
eigenvalues of R2 and Rz. The
operator R is defined by the equation
L⟩ transform according to the
analogous equations (eq. 33) and
(eq. 48), it is possible to use standard
vector coupling techniques [22] to obtain linear
combinations of products of the form |k′Jm⟩
| ′L⟩ which correspond to wave
functions characterized by the quantum numbers
|J m R kR⟩. The two new quantum
numbers R and kR, which replace k and
L, are
eigenvalues of R2 and Rz. The
operator R is defined by the equation
| (eq. 50) |
for s = 3 or 4. Molecule-fixed components of this equation are normally considered. It can be seen from (eq. 23) and (eq. 39) that all molecule-fixed components of J commute with all molecule-fixed components of Ls.
The rotational Hamiltonian for a spherical top molecule with zero or one quantum of a triply degenerate vibration excited can, to a first approximation, be written [4] as
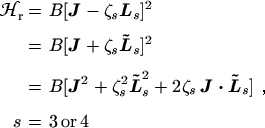 |
(eq. 51) |
corresponding qualitatively to the fact that the rotational energy of a
spherical top is given by the product of a rotational constant B, which
is inversely proportional to the moment-of-inertia of the molecule, and the
square of the purely rotational angular momentum
[J + ζs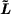 s]2.
Non-vanishing matrix elements of
s]2.
Non-vanishing matrix elements of 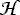 r are diagonal in the basis set
|J m R kR⟩, with values given by
[6]
r are diagonal in the basis set
|J m R kR⟩, with values given by
[6]
| (eq. 52) |
When  = 0,
= 0 and
R = J. When s = 1, s = 1 and
R = J + 1, J, or
J − 1. The Coriolis operator
+2B ζsJ · s
in (eq. 51) thus gives rise to a natural grouping of s = 1
rovibrational levels according to their R values. In
Fig. 5 the 3 = 1 levels are drawn in three
stacks, corresponding to JR = 67,
66, and 65. The relative energy positions of the stacks
are correct for a positive value of ζ3, but are not drawn
to scale.
= 0,
= 0 and
R = J. When s = 1, s = 1 and
R = J + 1, J, or
J − 1. The Coriolis operator
+2B ζsJ · s
in (eq. 51) thus gives rise to a natural grouping of s = 1
rovibrational levels according to their R values. In
Fig. 5 the 3 = 1 levels are drawn in three
stacks, corresponding to JR = 67,
66, and 65. The relative energy positions of the stacks
are correct for a positive value of ζ3, but are not drawn
to scale.
If laboratory-fixed components of the electric dipole moment operator are expanded as a power series in the vibrational coordinates Qs, and if only terms linear in these coordinates for one value of s are considered, we find
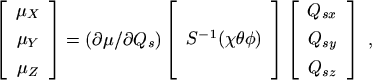 |
(eq. 53) |
where s = 3 or 4, and where (∂µ/∂Qs) represents any one of the three dipole derivatives (∂µx/∂Qsx), (∂µy/∂Qsy), or (∂µz/∂Qsz) evaluated at the equilibrium configuration. It can be shown by direct substitution that the right side of (eq. 53) commutes with the three components of the operator R given in (eq. 50). Consequently, to the approximation that only terms in the dipole moment expansion linear in the vibrational coordinates need be considered and to the approximation that R and kR are good quantum numbers, we obtain the well known [2] selection rules
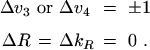 |
(eq. 54) |
These selection rules are valid for fundamental bands, but not for overtones and combination bands [59], whose intensity is governed by higher-order terms omitted in (eq. 53).
In Fig. 5, transitions indicated by solid lines
obey the selection rule ΔR = 0 and are strong. The five
Po(7) transitions and the one P−(7)
transition, indicated by dashed lines, violate this rule and are weaker. The
remaining weak vibration-rotation transition in Fig. 5, indicated by the
dashed line among the P+(7) transitions, does not violate the
ΔR = 0 selection rule. It does, however, violate a
selection rule requiring no change in the numerical counter superscript
[14,15]. This latter "selection rule"
depends on the relative values of the various interactions giving rise to the
splittings in the ground and first excited vibrational state [14,15], but has
been found to hold rather well for the infrared-active vibrational fundamentals
in CH4 [43−46,
61]. It is necessary, however, to "count"
the 3 = 1
rovibrational levels from highest energy to lowest when
R = J ± 1, and from lowest energy to highest
when R = J, to make this selection rule valid.
(Readers should be cautioned that slight variations of the numerical counting
scheme described here will be found in the methane literature, though all
schemes have in common the selection rule that the counting index does not
change for allowed transitions.)
![]()
|
| |
|
|
|